 Speeding Up Your Analytics with Machine Learning (Part 1) introduced how the use of deep learning (DL) greatly simplifies programming hybrid computers; and discussed data-driven programming. Part 2 introduces the use of DL to train a deep neural network (DNN) to further improve performance; and hybrid architectures.
Speeding Up Your Analytics with Machine Learning (Part 1) introduced how the use of deep learning (DL) greatly simplifies programming hybrid computers; and discussed data-driven programming. Part 2 introduces the use of DL to train a deep neural network (DNN) to further improve performance; and hybrid architectures.
Training and Inference
While we were searching for a simpler model than compute unified device architecture (CUDA) for data scientists, we realized that we could use DL to train a DNN. That gave us reasonable approximations of our analytics. Scoring with a DNN for this approximation, also called inference, gave us another order of magnitude of performance improvement for our analytics. Deep learning for numerical applications (DL4NA) was born.
We didn’t worry about the time it took for training, because we didn’t need to perform it very often.
Once we had DL4NA, we realized that many use cases were applicable for this methodology. Consider the use of DL4NA for approving loans over the phone or using a website. Many regulations govern the approval of a loan. For example, approving loans must be fair. You don’t want to explain to regulators that you approved the loan for Jane, but not for Sue, when they have almost identical financial risk profiles. Another very important consideration for banks is their overall risk profile (across all loans held by the bank). By approving this loan, does the bank expose itself to a higher risk? For example, does the bank have too many loans in one geographic area? The risk profile must be calculated across an entire portfolio of loans, not just for one loan at a time. So, for a bank with thousands or even millions of loans, this process of calculating the risk profile takes minutes, if not hours. This is clearly not desirable when you’re waiting in your browser for an approval. This problem is solved by DL4NA: you train a DNN to approximate your decision of approving or denying a loan application in a couple of seconds or less. Clearly, DL4NA makes the decision on a loan application fast, but it will probably not satisfy a regulator, since DL doesn’t easily give out its secrets. To meet regulatory requirements, the bank can run through its entire risk profile once at the end of the day, not every time a new loan application is considered.
Hybrid Architectures
In the example above, we used GPUs to run the training and the inference. Other devices are available to perform the same tasks.
Figure 1: Performance of Analytics with Hybrid Architectures
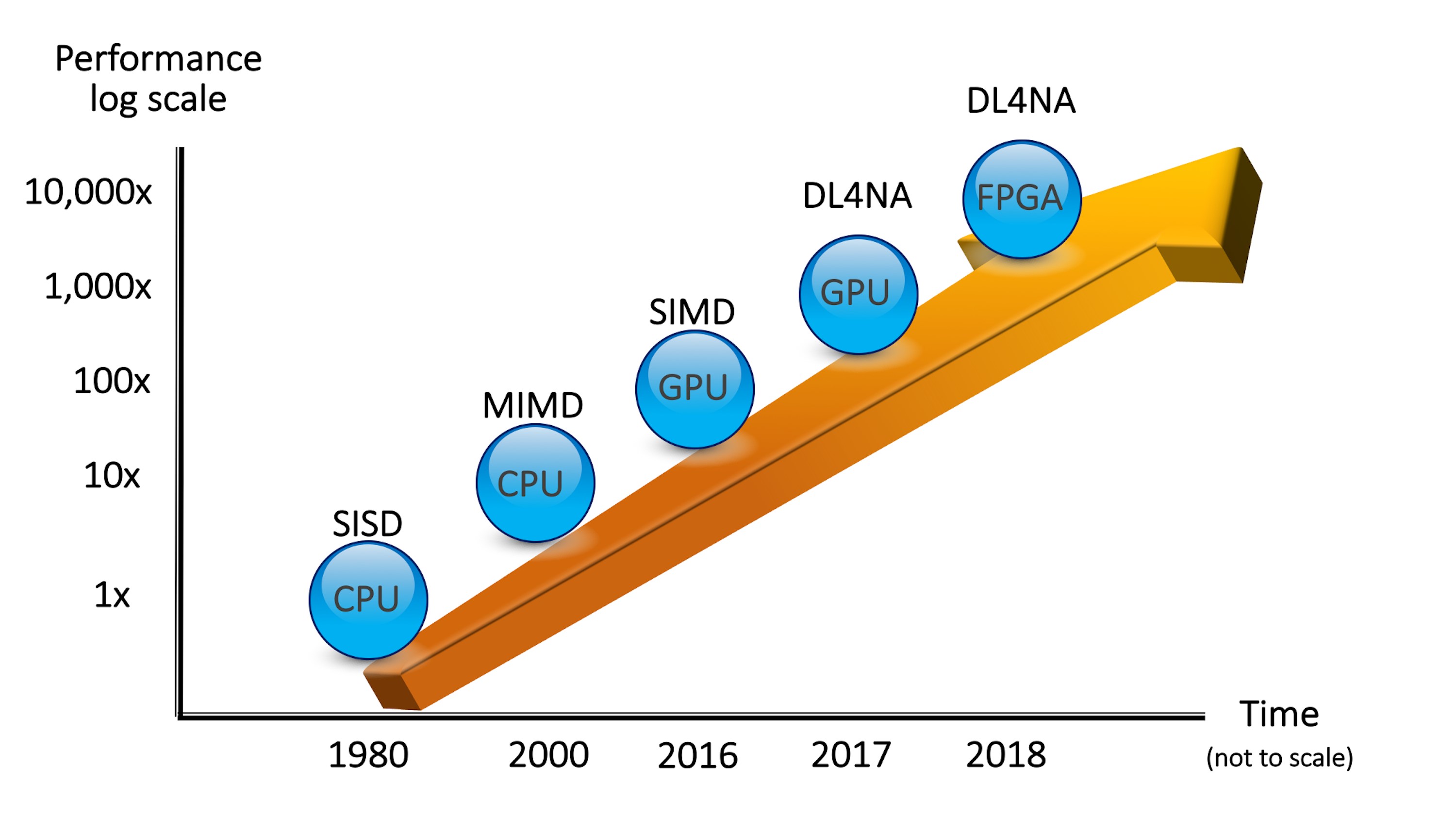
This marriage of CPUs and other devices is called a hybrid architecture. We believe that this is the way computers are going to be built in the future. This means that if you want to deploy your analytics so that they run as fast as possible, you must embrace hybrid architectures.
The good news is that with DL4NA, you are well-armed to deploy your analytics on hybrid architectures.
We should point out that even though the emphasis of this post has been on the great performance gains with hybrid architectures, you can also benefit from a wider platform to deploy your analytics. With DL4NA, you can deploy any analytics program that you can approximate using a DNN to any of those devices. In particular, you could run an approximation of a SAS or a Python program in an IoT device, thereby pushing your analytics to the edge of your network.
The content of this post is an excerpt of Chapter 9 of my book, Deep Learning for Numerical Applications with SAS®, where you will find an introduction to deep learning concepts in SAS along with step-by-step techniques that allow you to easily reproduce the examples on your high-performance analytics systems.
